
Search
- Page Path
- HOME > Search
Original Articles
- Hypothalamus and pituitary gland
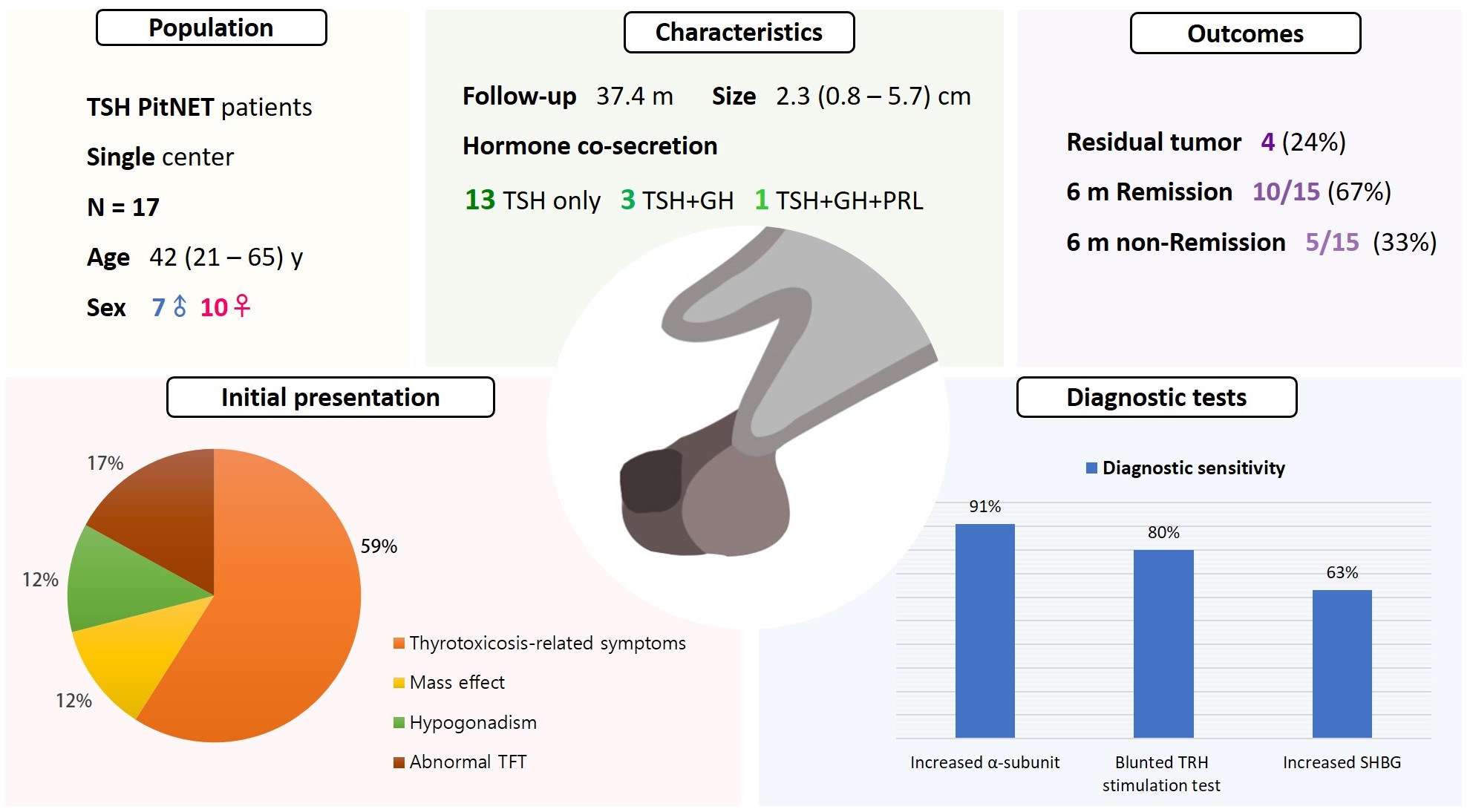
- Clinical Characteristics, Diagnosis, and Treatment of Thyroid Stimulating Hormone-Secreting Pituitary Neuroendocrine Tumor (TSH PitNET): A Single-Center Experience
- Jung Heo, Yeon-Lim Suh, Se Hoon Kim, Doo-Sik Kong, Do-Hyun Nam, Won-Jae Lee, Sung Tae Kim, Sang Duk Hong, Sujin Ryu, You-Bin Lee, Gyuri Kim, Sang-Man Jin, Jae Hyeon Kim, Kyu Yeon Hur
- Endocrinol Metab. 2024;39(2):387-396. Published online February 5, 2024
- DOI: https://doi.org/10.3803/EnM.2023.1877
- 1,437 View
- 39 Download
-
 Abstract
Abstract
 PDF
PDF Supplementary Material
Supplementary Material PubReader
PubReader  ePub
ePub - Background
Thyroid-stimulating hormone (TSH)-secreting pituitary neuroendocrine tumor (TSH PitNET) is a rare subtype of PitNET. We investigated the comprehensive characteristics and outcomes of TSH PitNET cases from a single medical center. Also, we compared diagnostic methods to determine which showed superior sensitivity.
Methods
A total of 17 patients diagnosed with TSH PitNET after surgery between 2002 and 2022 in Samsung Medical Center was retrospectively reviewed. Data on comprehensive characteristics and treatment outcomes were collected. The sensitivities of diagnostic methods were compared.
Results
Seven were male (41%), and the median age at diagnosis was 42 years (range, 21 to 65); the median follow-up duration was 37.4 months. The most common (59%) initial presentation was hyperthyroidism-related symptoms. Hormonal co-secretion was present in four (23%) patients. Elevated serum alpha-subunit (α-SU) showed the greatest diagnostic sensitivity (91%), followed by blunted response at thyrotropin-releasing hormone (TRH) stimulation (80%) and elevated sex hormone binding globulin (63%). Fourteen (82%) patients had macroadenoma, and a specimen of one patient with heavy calcification was negative for TSH. Among 15 patients who were followed up for more than 6 months, 10 (67%) achieved hormonal and structural remission within 6 months postoperatively. A case of growth hormone (GH)/TSH/prolactin (PRL) co-secreting mixed gangliocytoma-pituitary adenoma (MGPA) was discovered.
Conclusion
The majority of the TSH PitNET cases was macroadenoma, and 23% showed hormone co-secretion. A rare case of GH/TSH/PRL co-secreting MGPA was discovered. Serum α-SU and TRH stimulation tests showed great diagnostic sensitivity. Careful consideration is needed in diagnosing TSH PitNET. Achieving remission requires complete tumor resection. In case of nonremission, radiotherapy or medical therapy can improve the long-term remission rate.

- Clinical Study
- Associations of GNAS Mutations with Surgical Outcomes in Patients with Growth Hormone-Secreting Pituitary Adenoma
- Hyein Jung, Kyungwon Kim, Daham Kim, Ju Hyung Moon, Eui Hyun Kim, Se Hoon Kim, Cheol Ryong Ku, Eun Jig Lee
- Endocrinol Metab. 2021;36(2):342-350. Published online March 23, 2021
- DOI: https://doi.org/10.3803/EnM.2020.875

- 4,370 View
- 144 Download
- 4 Web of Science
- 5 Crossref
-
Abstract
PDFPubReader ePub
- Background
The guanine nucleotide-binding protein, alpha stimulating (GNAS) gene has been associated with growth hormone (GH)-secreting pituitary adenoma. We investigated the prevalence of GNAS mutations in Korean patients with acromegaly and assessed whether mutation status correlated with biochemical or clinical characteristics.
Methods
We studied 126 patients with acromegaly who underwent surgery between 2005 and 2014 at Severance Hospital. We performed GNAS gene analysis and evaluated age, sex, hormone levels, postoperative biochemical remission, and immunohistochemical staining results of the tumor.
Results
GNAS mutations were present in 75 patients (59.5%). Patients with and without GNAS mutations showed similar age distribution and Knosp classification. The proportion of female patients was 76.5% and 48.0% in the GNAS-negative and GNAS-mutation groups, respectively (P=0.006). In immunohistochemical staining, the GNAS-mutation group showed higher GH expression in pituitary tumor tissues than the mutation-negative group (98.7% vs. 92.2%, P=0.015). Patients with GNAS mutations had higher preoperative insulin-like growth factor-1 levels (791.3 ng/mL vs. 697.0 ng/mL, P=0.045) and lower immediate postoperative basal (0.9 ng/mL vs. 1.0 ng/mL, P=0.191) and nadir GH levels (0.3 ng/mL vs. 0.6 ng/mL, P=0.012) in oral glucose tolerance tests. Finally, the GNAS-mutation group showed significantly higher surgical remission rates than the mutation-negative group, both at 1 week and 6 months after surgical resection (70.7% vs. 54.9%, P=0.011; 85.3% vs. 82.4%, P=0.007, respectively).
Conclusion
GNAS mutations in GH-secreting pituitary tumors are associated with higher preoperative insulin-like growth factor-1 levels and surgical remission rates and lower immediate postoperative nadir GH levels. Thus, GNAS mutation status can predict surgical responsiveness in patients with acromegaly. -
Citations
Citations to this article as recorded by
- Genetic diagnosis in acromegaly and gigantism: From research to clinical practice
Claudia Ramírez-Rentería, Laura C. Hernández-Ramírez
Best Practice & Research Clinical Endocrinology & Metabolism.2024; : 101892. CrossRef - CD8/PD-L1 immunohistochemical reactivity and gene alterations in cutaneous squamous cell carcinoma
Haruto Nishida, Yoshihiko Kondo, Takahiro Kusaba, Kazuhiro Kawamura, Yuzo Oyama, Tsutomu Daa, Avaniyapuram Kannan Murugan
PLOS ONE.2023; 18(2): e0281647. CrossRef - Dynamic monitoring of circulating tumor DNA to analyze genetic characteristics and resistance profile of lorlatinib in ALK positive previously treated NSCLC
Xiya Ma, Kun Zhang, Jing Xu, Hongjun Gao, Shaoxing Yang, Haifeng Qin, Hong Wang, Fang Gao, Xiaoqing Liu
Thoracic Cancer.2023; 14(20): 1980. CrossRef - Multiomics Approach to Acromegaly: Unveiling Translational Insights for Precision Medicine
Kyungwon Kim, Cheol Ryong Ku, Eun Jig Lee
Endocrinology and Metabolism.2023; 38(5): 463. CrossRef - Hotspots of Somatic Genetic Variation in Pituitary Neuroendocrine Tumors
Mariana Torres-Morán, Alexa L. Franco-Álvarez, Rosa G. Rebollar-Vega, Laura C. Hernández-Ramírez
Cancers.2023; 15(23): 5685. CrossRef
- Genetic diagnosis in acromegaly and gigantism: From research to clinical practice
- Genetic and Epigenetic Analysis in Korean Patients with Multiple Endocrine Neoplasia Type 1
- Yoon Jung Chung, Sena Hwang, Jong Ju Jeong, Sun Yong Song, Se Hoon Kim, Yumie Rhee
- Endocrinol Metab. 2014;29(3):270-279. Published online September 25, 2014
- DOI: https://doi.org/10.3803/EnM.2014.29.3.270
- 3,603 View
- 49 Download
- 3 Web of Science
- 4 Crossref
-
Abstract
PDFPubReader
Background Multiple endocrine neoplasia type 1 (MEN1) is a familial syndrome characterized by the parathyroid, pancreas and pituitary tumors. Parathyroid tumors are the most common clinical manifestations, occurring in more than 90% of MEN1 patients. Heterozygous germline mutations of the
MENIN gene underlie the tumorigenesis in MEN1 and epigenetic alterations along with germline mutations may contribute to tumorigenesis. Here, we investigated the associations between genotype and phenotype in Korean MEN1 patients.Methods We analyzed medical records from 14 unrelated MEN1 patients who had newly confirmed
MENIN germline mutations, together with 14 previous reports in Korea. Aberrant DNA methylations were also examined in MEN1-related parathyroid tumors using the Infinium HumanMethylation 450 BeadChip.Results Total 28 germline mutations of
MENIN were relatively highly concentrated in exons 7 and 8 compared to previous reports from Western countries. Six mutations (c.111dupT/p.S38Ffs*79, c.225_226insT/p.T76Yfs*41, c.383_398del16/p.S128Tfs*52, c.746dupT/p.H250Afs*20, c.1150G>T/p.E384*, and c.1508G>A/p.G503N) were newly found in the present study. Of interest, four patients (15%) showed unusual initial presentations and three patients were diagnosed incidentally at the general medical checkup. We also found three distinct sites in exon 2 ofMENIN were significantly hypomethylated in the MEN1 parathyroid tumors, comparing correspondent blood samples.Conclusion We also have found a lack of genotype/phenotype correlation in Korean MEN1 patients. There were not a few unusual initial manifestations in MEN1 patients, thus, genetic testing for the
MENIN germline mutations can provide important information for the better prognosis. Further studies are warranted to investigate altered DNA methylations in theMENIN gene involved in tumorigenesis.-
Citations
Citations to this article as recorded by- Cutaneous lesions and other non-endocrine manifestations of Multiple Endocrine Neoplasia type 1 syndrome
Laura Pierotti, Elena Pardi, Elisa Dinoi, Paolo Piaggi, Simona Borsari, Simone Della Valentina, Chiara Sardella, Angela Michelucci, Maria Adelaide Caligo, Fausto Bogazzi, Claudio Marcocci, Filomena Cetani
Frontiers in Endocrinology.2023;[Epub] CrossRef - A Case of Asymptomatic Multiple Endocrine Neoplasia Type I with Thymic Carcinoid
Suk Ki Park, Moon Won Lee, In Sub Han, Young Joo Park, Sung Yong Han, Joon Woo Park, Bong Eun Lee, Gwang Ha Kim, Sang Soo Kim
The Korean Journal of Helicobacter and Upper Gastrointestinal Research.2019; 19(1): 65. CrossRef - Multiple Endocrine Neoplasia Syndromes from Genetic and Epigenetic Perspectives
Fatemeh Khatami, Seyed Mohammad Tavangar
Biomarker Insights.2018; 13: 117727191878512. CrossRef - Articles in 'Endocrinology and Metabolism' in 2014
Won-Young Lee
Endocrinology and Metabolism.2015; 30(1): 47. CrossRef
- Cutaneous lesions and other non-endocrine manifestations of Multiple Endocrine Neoplasia type 1 syndrome
 First
First Prev
Prev


